Zespół Blooma: objawy, przyczyny i leczenie
Zespół Blooma (BS) jest rzadką chorobą dziedziczenia autosomalnego recesywnego, charakteryzującą się głównie trzema aspektami: opóźnieniem wzrostu, nadwrażliwością na słońce i teleangiektazjami na twarzy (rozszerzenie naczyń włosowatych). Pacjenci ci mają niestabilność genomiczną, która predysponuje ich do łatwego rozwoju raka.
Został odkryty przez dermatologa Davida Blooma w 1954 r. Dzięki obserwacji kilku pacjentów, którzy prezentowali karłowatość i rumień naczynioruchowy (zaczerwieniona skóra z powodu rozszerzenia naczyń włosowatych) (Elbendary, 2015).
Zespół ten można również nazwać wrodzonym rumieniem telangiektatycznym lub zespołem Blooma-Torre-Machacka.
Przyczyny zespołu Blooma
Zespół Blooma jest chorobą autosomalną recesywną, to znaczy mutacja musi wystąpić w obu allelach genu BLM, zarówno od matki, jak i od ojca (Ellis i in., 1995). Rodzice niekoniecznie muszą mieć tę chorobę, ale mogą być nosicielami zmutowanego genu bez objawów.
W genie BLM w zespole Blooma znaleziono ponad 60 mutacji, z których najczęstszą jest delecja 6 nukleotydów w pozycji 2281 i podstawienie przez 7 innych (Elbendary, 2015).
Według Genetics Home Reference (2016) gen BLM odpowiada za wysyłanie instrukcji dotyczących tworzenia białka RecQ, które jest częścią rodziny helikaz.
To, co helikazy robią, to połączenie DNA i tymczasowe oddzielenie dwóch nici, które są zwykle połączone w spiralę, w celu opracowania procesów takich jak replikacja (lub kopia DNA), przygotowanie do podziału komórki i naprawy. uszkodzeń DNA.
Krótko mówiąc, helikazy RecQ są ważne dla utrzymania struktury DNA i dlatego są znane jako „opiekunowie genomu”.
Na przykład, kiedy komórka ma się podzielić, tworząc dwie nowe komórki, DNA znajdujące się w chromosomach musi zostać skopiowane, tak aby każda nowa komórka miała dwie kopie każdego chromosomu: jednego z ojca i drugiego z matki.
DNA skopiowane z każdego chromosomu ma dwie identyczne struktury, które nazywane są chromatydami siostrzanymi i są połączone na początku, zanim nastąpi podział komórek.
Na tym etapie wymieniają między sobą fragmenty DNA; co jest znane jako wymiana chromatyd siostrzanych. Wydaje się, że ten proces jest zmieniony w chorobie Blooma, ponieważ białko BLM jest uszkodzone i to kontroluje prawidłową wymianę między chromatydami siostrzanymi i że DNA pozostaje stabilne w czasie kopiowania.
W rzeczywistości istnieje średnio 10 więcej niż normalna wymiana między chromatydami w zespole Blooma (Seki et al., 2006).
Z drugiej strony, w tej chorobie dochodzi do przerw w materiale genetycznym, które powodują pogorszenie normalnej aktywności komórkowej, której ze względu na brak białka BLM nie można naprawić.
W rzeczywistości niektórzy eksperci klasyfikują ten zespół jako „zespół złamania chromosomów”, ponieważ wiąże się on z dużą liczbą przerw i rearanżacji chromosomów.
Ta niestabilność chromosomów powoduje większe prawdopodobieństwo rozwoju chorób. Na przykład, z powodu braku białka BLM, nie mogą one powrócić do zdrowia po uszkodzeniu DNA, które może powodować światło ultrafioletowe, a zatem pacjenci ci są wrażliwi na światło.
Ponadto osoby dotknięte chorobą mają niedobór odporności, który czyni je bardziej podatnymi na infekcje.
Z drugiej strony, mają wysokie prawdopodobieństwo zachorowania na raka w dowolnym narządzie przez niekontrolowany podział komórek, pojawiający się głównie w białaczce (jest to rodzaj nowotworu krwi charakteryzującego się nadmiarem białych krwinek) i chłoniakiem (rak w węźle chłonnym układu). odporny).
Awarie znaleziono również w działaniu genu FANCM, który jest odpowiedzialny za kodowanie białek MM1 i MM2, które również służą do naprawy uszkodzeń DNA.
Są to te, które były związane zarówno z tym zespołem, jak i z niedokrwistością Fanconiego. Dlatego widzimy, że te dwie choroby są podobne pod względem fenotypu i predyspozycji do guzów hematologicznych i niewydolności szpiku kostnego.
W każdym razie mechanizmy molekularne, które wpływają na chromosomy w zespole Blooma, są nadal badane.
Jaka jest jego częstość?
Zespół Blooma jest stosunkowo rzadki, znanych jest tylko około 300 przypadków opisanych w literaturze medycznej. Chociaż zaburzenie to występuje w wielu grupach etnicznych, wydaje się, że jest ono znacznie częstsze u Żydów aszkenazyjskich, stanowiąc 25% pacjentów z tym zespołem.
W rzeczywistości w tej grupie etnicznej częstość występowania zespołu może osiągnąć 1%. Stwierdzono również, choć rzadziej, w rodzinach japońskich.
Jeśli chodzi o seks, mężczyźni wydają się być bardziej narażeni na chorobę niż kobiety, przy czym odsetek ten wynosi 1, 3 mężczyźni na 1 kobietę.
Jakie są jego objawy?
Ten stan występuje już w pierwszych miesiącach życia i na razie żaden z pacjentów nie przeżył ponad 50 lat.
- Nowotwory złośliwe : spowodowane niestabilnością genomową, jak wyjaśniono powyżej, są główną przyczyną śmierci osób dotkniętych tym zespołem. Według National Organization for Rare Disorders (2014) około 20% osób dotkniętych zespołem Blooma będzie miało raka. Pacjenci ci mają od 150 do 300 razy większe ryzyko zachorowania na raka niż osoby bez tego zaburzenia.
- Niedobór odporności, który zmienia się w zależności od pacjenta i predysponuje do różnych infekcji. Wynika to z deficytów w proliferacji limfocytów (białych krwinek), problemów w syntezie immunoglobulin (przeciwciał układu odpornościowego) i niskiej odpowiedzi na stymulację przez mitogeny (które kontrolują podział i wzrost komórek).
- Wady limfocytów T i B są częste, wpływając na rozwój układu odpornościowego.
- Wadliwe działanie układu odpornościowego może spowodować infekcję ucha (głównie zapalenie ucha środkowego), zapalenie płuc lub inne objawy, takie jak biegunka i wymioty.
- Nadwrażliwość na światło : to nadmierna wrażliwość DNA na promienie ultrafioletowe, która ulega uszkodzeniu. Uważa się, że jest to forma fototoksyczności lub śmierci komórki, która szkodzi skórze dotkniętego chorobą, gdy słońce uderza.
- Zmniejszenie płodności lub niepłodności . W rzeczywistości u mężczyzn nie ma możliwości oczekiwania. U kobiet występuje bardzo wczesna menopauza.
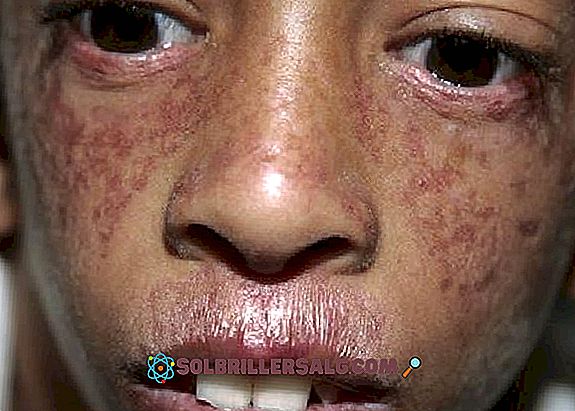
- Objawy skórne : oprócz nadwrażliwości na światło, występuje również poikiloderma, która dotyka skóry, pojawia się głównie w strefie hipopigmentacji, innych przebarwieniach, teleangiektazjach i atrofii. Często obserwuje się czerwone plamy na skórze związane z ekspozycją na słońce (szczególnie na twarzy).
- Kolejnym obserwowanym problemem skórnym jest teleangiektazja, która jest postrzegana jako czerwonawe wykwity na twarzy spowodowane rozszerzeniem małych naczyń krwionośnych. Pojawia się jako wzór „motyla” pokrywający nos i policzki.
- Nieprawidłowe brązowe lub szare plamy mogą również pojawić się na innych częściach ciała (plamy „café au lait”).
- Opóźnienie w rozwoju, który już objawia się u niemowląt. Najmłodsze mają zwykle charakterystyczną głowę i twarz, węższe i mniejsze niż normalnie.
- Około 10% dotkniętych chorobą rozwija się w cukrzycy (National Organization for Rare Disorders, 2014).
- Bardzo ostry głos .
- Zmiany w zębach .
- Nieprawidłowości w oczach, uszach (wydatne uszy), dłoniach lub stopach (takie jak polidaktylia, które występują, gdy pacjent ma więcej palców niż normalnie).
- Torbiele pilonidalne.
- Problemy z karmieniem : są zauważane zwłaszcza u niemowląt i małych dzieci, co wskazuje na brak zainteresowania jedzeniem. Towarzyszy temu wiele razy ciężki refluks żołądkowo-przełykowy.
- Zdolności intelektualne są zmienne, tak że u niektórych pacjentów są bardziej zniszczone, aw innych są w normie.
Jak jest diagnozowana?
Można go zdiagnozować za pomocą jednego z następujących testów:
- Testy cytogenetyczne mierzące aberracje chromosomalne i poziom wymiany chromatyd siostrzanych.
Możliwe jest zaobserwowanie obecności cztero-promieniowych powiązań (wymiana chromatyd czteroramiennych) w limfocytach hodowanych we krwi, w celu sprawdzenia, czy występują wysokie poziomy wymiany chromatyd siostrzanych w dowolnej komórce, przerwy chromatydowe, przerwy lub przegrupowania; Lub zobacz bezpośrednio, czy są mutacje w genie BLM.
Testy te mogą wykryć zdrowego osobnika, który nosi mutacje w genie BLM i może przekazać je swojemu potomstwu.
Amerykańska Agencja ds. Żywności i Leków (FDA) ogłosiła w lutym 2015 r. Komercjalizację testu genetycznego „23andMe”, który może być przydatny do wczesnego wykrywania tej choroby.
Obecność tego zespołu należy podejrzewać, jeśli istnieją takie warunki kliniczne:
- Opóźnienie znacznego wzrostu obserwowanego od okresu wewnątrzmacicznego.
- Obecność rumieni na skórze twarzy po ekspozycji na słońce.
Nie myl z ...
Przed zdiagnozowaniem zespołu Blooma należy wziąć pod uwagę następujące syndromy:
- Inne zespoły autosomalnej recesywnej niestabilności chromosomalnej, które są związane z przerwami i rearanżacjami chromosomów, powodując, że pacjent jest szczególnie podatny na pewne typy nowotworów, takie jak: niedokrwistość Fanconiego, ataksja teleangiektazja lub pigmentacja kseroderma, które obejmują inne geny, a nie BLM,
- Zespół Cockayne'a, na który składa się zaburzenie dziedziczne objawiające się opóźnionym rozwojem, nadwrażliwością na światło i starzeniem się w młodym wieku. Jest to rzadka forma karłowatości.
- Zespół Rothmunda-Thomsona : niezwykle rzadki i objawiający się typowymi nieprawidłowościami skóry, wadami włosów, młodzieńczą zaćmą, niedoborem wzrostu i zmianami szkieletowymi, takimi jak wady rozwojowe twarzoczaszki. Przypomina zespół Blooma w zapaleniu skóry, poikilodermie, zwyrodnieniu skóry (atrofii) i teleangiektazjach.
Leczenie
Nie ma specyficznego leczenia zespołu Blooma, czyli nadmiernej liczby mutacji. Interwencje mają raczej na celu złagodzenie objawów, oferowanie wsparcia i zapobieganie powikłaniom.
- Staraj się nie narażać bezpośrednio na słońce.
- Użyj odpowiedniego kremu do opalania.
- Kontrola dermatologa w celu leczenia plam, zaczerwienienia i stanu zapalnego skóry.
- Używaj antybiotyków do infekcji.
- Okresowe kontrole lekarskie w celu wykrycia ewentualnych przypadków raka, głównie wtedy, gdy pacjenci osiągają dorosłość. Musimy starać się zwracać uwagę na możliwe objawy, ponieważ istnieją guzy wymagające wczesnego usunięcia chirurgicznego w celu ich powrotu do zdrowia. Niektóre metody wczesnego diagnozowania raka to mammografia, cytologia Papanicolaou lub dopochwowa lub kolonoskopia.
- Kontroluj, czy te dzieci otrzymują niezbędne składniki odżywcze, próbując interweniować w refluksie trawiennym. W tym celu można umieścić rurkę w górnej części przewodu pokarmowego w celu uzupełnienia karmienia podczas snu. To może trochę zwiększyć ilość złogów tłuszczu, ale nie wydaje się mieć wpływu na sam wzrost.
- Zbadaj istnienie cukrzycy, aby ją leczyć jak najszybciej.
- Jeśli osoba ma raka, można rozważyć przeszczep szpiku kostnego.
- Rodzinne i inne grupy wsparcia oraz stowarzyszenia o podobnych chorobach, tak aby dotknięta chorobą osoba rozwijała się jako osoba o możliwie najwyższej jakości życia.
- Jeśli rodzina miała przypadki tej choroby lub przez rodzinę małżonka, poradnictwo genetyczne byłoby przydatne w celu uzyskania informacji na temat natury, dziedziczenia i konsekwencji tego rodzaju zaburzeń, aby przyczynić się do podejmowania decyzji medycznych i osobiste.