Zespół Zellwegera: objawy, przyczyny, leczenie
Zespół Zellwegera, znany również jako zespół Cerebro-Hepato-Renal, jest rodzajem patologii metabolicznej klasyfikowanej w ramach chorób peroksysomalnych (Cáceres-Marzal, Vaquerizo-Madrid, Girós, Ruiz i Roels, 2003).
Choroba ta charakteryzuje się nieprawidłowym przetwarzaniem lub gromadzeniem różnych związków, kwasu fitanowego, cholesterolu lub kwasów żółciowych w różnych obszarach, takich jak mózg, wątroba lub nerki (szpital Sant Joan de Déu, 2016).

Klinicznie, zespół Zellwegera jest definiowany przez prezentację różnych medycznych objawów i symptomów związanych z anomaliami i deformacjami twarzy, hepatomegalią i ciężką dysfunkcją neurologiczną (Rodillo i in., 1996).
Ponadto etiologiczne pochodzenie tej choroby stwierdza się w anomalii genetycznej, która powoduje niedobór produkcji lub aktywności peroksysomów. Składnik komórkowy o znaczącej roli w metabolizmie różnych substancji biochemicznych w naszym organizmie (Girós, López Pisón, Luisa Serrano, Sierra, Toledo i Pérez-Cerdá, 2016).
W odniesieniu do diagnozy zespołu Zellwegera, oprócz badania fizykalnego i identyfikacji objawów klinicznych, obejmuje on szeroki zakres testów laboratoryjnych: analizę biochemiczną, badania histologiczne, neuroobrazowanie, ultradźwięki, elektroencefalografię, badania okulistyczne, analizę funkcja serca itp. (Cáceres-Marzal, Vaquerizo-Madryt, Girós, Ruiz i Roels, 2003).
Badania eksperymentalne w toku nie zdołały jeszcze zidentyfikować leku na zespół Zellwegera. Wszystkie interwencje terapeutyczne opierają się głównie na leczeniu objawowym i paliatywnym (szpital Sant Joan de Déu, 2016).
W większości przypadków osoby dotknięte zespołem Zellwegera zwykle nie przekraczają 2 lat życia z powodu istotnych powikłań medycznych.
Charakterystyka zespołu Zellwegera
Zespół Zellwegera jest wrodzoną patologią pochodzenia genetycznego, klasyfikowaną w ramach tak zwanych chorób peroksysomalnych lub zaburzeń biogenezy peroksysomów (National Institute of Neurological Disorders and Stroke, 2016).
Zaburzenia lub choroby peroksysomalne stanowią szeroką grupę patologii metabolicznych spowodowanych nieprawidłowością w tworzeniu lub funkcjonowaniu peroksysomów (szpital Sant Joan de Déu, 2016):
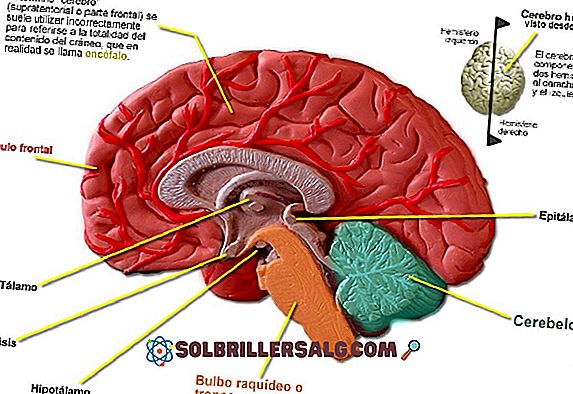
Peroksysom jest organellą komórkową, która zawiera w swoim wnętrzu różne białka odpowiedzialne za realizację licznych funkcji metabolicznych, takich jak degradacja lub synteza substancji biochemicznych.
Ten organelle lub związek komórkowy może znajdować się w prawie wszystkich tkankach organizmu, jednak częściej występuje w obszarach mózgu, nerek lub wątroby.
Ponadto mogą być one tworzone przez podział i namnażanie istniejących komórek lub przez nowe procesy proliferacji, syntetyzowane przez różne białka zlokalizowane w jądrach komórkowych.
Dlatego na biogenezę lub wytwarzanie peroksysomu wpływa aktywność różnych białek, kodowanych na poziomie genetycznym przez około 16 różnych genów, których zmiana może powodować istotne anomalie w tym związku komórkowym.
W przypadku zespołu Zellwegera występuje nieprawidłowość w biogenezie peroksysomu, która powoduje patologiczne nagromadzenie różnych związków, które są toksyczne lub nie zostały prawidłowo zdegradowane.
Analizy biochemiczne przeprowadzone w dziedzinie badań zespołu Zellwegera wykazały wysokie stężenia kwasu fitanowego, kwasów wielonienasyconych, kwasów w cholesterolu moczu, kwasów żółciowych itp.
Ponadto tego typu zmiany mogą również mieć znaczący wpływ na syntezę plazmogenów, podstawowej substancji w rozwoju mózgu.
W konsekwencji osoby dotknięte zespołem Zellwegera wykazują wiele różnych objawów neurologicznych, nieprawidłowości czaszkowo-twarzowych, zmiany w nerkach i wątrobie, które poważnie zagrażają ich przetrwaniu (Cáceres-Marzal, Vaquerizo-Madryt, Girós, Ruiz i Roels)., 2003).
Choroba ta została początkowo opisana przez Hansa Zellwegera (1964), od którego otrzymał nazwisko i grupę roboczą (Valdez Geraldo i in., 2009).
W swoim raporcie klinicznym Zellweger odniósł się do dwóch pacjentów z rodzeństwem, których stan kliniczny charakteryzował się niepowodzeniem wielofunkcyjnym, związanym z brakiem peroksysomów.
Następnie w 1973 r. Goldfischer i współpracownicy zlokalizowali nieobecność tych organelli komórkowych na określonym poziomie w nerkach i wątrobie (Prudencio Beltrán, Coria Miranda, Nubela Salguero, Pemintel Zárate, 2009).
Obecnie zespół Zellwegera definiuje się jako jeden z najpoważniejszych wariantów chorób peroksysomalnych, których charakterystyka kliniczna spowoduje systematyczne pogorszenie osoby dotkniętej chorobą (Braverman, 2012).
Statystyki
Zespół Zellwegera uważany jest za rzadką patologię, rzadką w populacji ogólnej (Genetics Home Reference, 2016).
Badania statystyczne wykazały przybliżoną częstość występowania jednego przypadku na 50 000 osób (Genetics Home Reference, 2016).
Jeśli chodzi o cechy socjodemograficzne związane z występowaniem tej choroby, obecne badania nie wykazały większej częstości występowania związanej z płcią, pochodzeniem geograficznym lub przynależnością do poszczególnych grup etnicznych i / lub rasowych (Krajowa Organizacja Rzadkich Zaburzeń, 2013 ).
Mimo to niektórzy autorzy, tacy jak (Braverman, 2012), zwracają uwagę na obecność zróżnicowanego rozpowszechnienia związanego z różnymi krajami:
- Stany Zjednoczone: 1 przypadek na każde 50 000 mieszkańców.
- Japonia: 1 przypadek na 500 000 mieszkańców.
- Suguenay-Lac Sant Jean (Quebec): 1 przypadek na każde 12 000 mieszkańców.
Ponadto, w wielu przypadkach ta patologia pozostaje nierozpoznana z powodu jej szybkiego postępu i wysokiej śmiertelności, tak że dane statystyczne dotyczące jej występowania mogą być niedoszacowane (National Organization for Rare Disorders, 2013).
Znaki i objawy
Charakterystyka kliniczna zespołu Zellwegera zostanie podzielona na kilka grup: zmiany twarzoczaszki, zmiany neurologiczne i anomalie wątrobowe / nerkowe (Genetics Home Reference, 2016, National Organization for Rare Disorders, 2013).
Zaburzenia twarzoczaszki
Wiele osób z zespołem Zellwegera ma atypowy fenotyp twarzy charakteryzujący się:
- Dolichocephaly : globalna struktura czaszki może mieć nieprawidłową strukturę, charakteryzującą się wydłużeniem regionów przednich i tylnych.
- Spłaszczony wygląd twarzy : ogólnie rzecz biorąc, wszystkie struktury, które tworzą twarz, zazwyczaj wykazują słabe rozwinięcie. W tym sensie wydają się być mniejsze niż normalnie lub, przeciwnie, rozwijają się niecałkowicie. W rezultacie wrażenie wizualne jest spłaszczeniem obszaru twarzy.
- Płaski potyliczny: tył głowy, znajdujący się pomiędzy ostatnią częścią czaszki a szyją, może słabo rozwinąć się, powodując nienormalnie spłaszczoną konfigurację.
- Wyłupiaste, szerokie i szerokie czoło: na ogół całkowity rozmiar czoła lub czołowej części czaszki jest zwykle większy niż normalnie, co wskazuje na znaczny występ.
- Szeroki korzeń nosa: struktura kości nosa zwykle rozwija się z objętością większą niż normalna lub oczekiwana, więc zazwyczaj ma szeroki i solidny wygląd. Ponadto obecność odwróconych kanałów nosowych jest zwykle kolejną z najczęstszych cech tego zespołu.
- Anomalie okulistyczne: doły oczne są zwykle źle zdefiniowane. Ponadto często rozwija się patologia rogówki. Wiele osób dotkniętych chorobą może mieć znacznie zmniejszoną zdolność widzenia.
- Micrognathia: w tym przypadku zarówno podbródek, jak i reszta struktury szczęki mają tendencję do rozwijania się ze zmniejszoną objętością, co powoduje inne zmiany w jamie ustnej i wtórne.
- Wady rozwojowe szpilki słuchowej : uszy zwykle wydają się zniekształcone lub z bardzo słabym rozwojem. W tym sensie nie tylko powodują zniekształcenia estetyczne, ale mogą wystąpić liczne przypadki ograniczonego słuchu.
- Zaburzenia jamy ustnej: w przypadku wewnętrznej struktury jamy ustnej często obserwuje się rozszczep podniebienia.
- Nadmiar skóry : w szczególności często stwierdza się znaczny nadmiar skóry na szyi.
Zmiany neurologiczne
Patologie związane ze strukturą i funkcją układu nerwowego są zwykle najpoważniejszymi objawami zespołu Zellwegera.
Ogólnie, neurologiczne komplikacje medyczne są głównie spowodowane zmianą migracji neuronów, utratą lub uszkodzeniem osłonek mielinowych (demielinizacja) i znaczącą atrofią istoty białej (leukodystrofia).
W związku z tym można również obserwować rozwój makrocefalii (nieprawidłowy wzrost obwodu czaszki) lub mikrocefalii (znaczące zmniejszenie obwodu czaszki).
Dlatego niektóre wtórne powikłania tych zmian neurologicznych charakteryzują się obecnością:
- Napady padaczkowe: anomalie strukturalne i funkcjonalne na poziomie mózgu mogą powodować niedobór i asynchroniczną neuronalną aktywność elektryczną. Dlatego może prowadzić do nawracających epizodów nagłych i niekontrolowanych skurczów mięśni, sztywności mięśni lub okresów nieobecności.
- Hipotonia mięśni : na ogół grupy mięśni mają tendencję do zmniejszonego i niefunkcjonalnego tonu, co utrudnia wykonywanie jakiejkolwiek aktywności ruchowej.
- Słuch i utrata wzroku : oprócz wad rozwojowych addytywnych i okulistycznych, możliwe jest, że nastąpiła zmiana zdolności widzenia i słuchu wtórna do anomalii neurologicznych, takich jak uszkodzenie zakończeń nerwów obwodowych.
- Niepełnosprawność intelektualna : wiele anomalii neurologicznych zwykle wiąże się z bardzo ograniczonym rozwojem intelektualnym i poznawczym.
Wady wątroby i nerek
Pomimo mniejszej częstości występowania, w porównaniu z opisanymi powyżej objawami, niektóre układy, takie jak nerka lub wątroba, mogą być znacząco osłabione:
- Splenomegalia : śledziona i sąsiednie struktury mogą rosnąć bardziej niż zwykle, powodując różne anomalie funkcjonalne.
- Hepatomegalia : wątroba zwykle rozwija się nieprawidłowo, osiągając większy rozmiar normalny lub podtrzymywany przez strukturę ciała.
- Marskość wątroby : ze względu na zmiany metaboliczne możliwe jest, że w wątrobie dochodzi do nieprawidłowego i patologicznego przechowywania materiału tłuszczowego.
- Żółtaczka : podobnie jak w innych przypadkach, niedobory metaboliczne mogą prowadzić do obecności nieprawidłowo wysokich poziomów bilirubiny we krwi, powodując żółte zabarwienie na poziomie skóry i oczu.
Przyczyny
Jak wskazaliśmy w początkowym opisie, zespół Zellwegera ma swoje źródło w niedostatecznej biogenezie peroksoomii (Girós, López Pisón, Luisa Serrano, Sierra, Toledo i Pérez-Cerdá, 2016).
Jednak ten nieprawidłowy mechanizm metaboliczny znajduje przyczynę etiologiczną w zmianie genetycznej.
Konkretnie, różne badania miały na celu zidentyfikowanie specyficznych mutacji w wielu różnych genach, około 14-16 (Girós, López Pisón, Luisa Serrano, Sierra, Toledo i Pérez-Cerdá, 2016).
Chociaż nie wszystkie funkcje tych genów są dokładnie znane, zaobserwowano, że odgrywają one znaczącą rolę w generowaniu instrukcji biochemicznych do produkcji grupy białek zwanych peroksynami (Genetics Home Reference, 2016).
Ten typ białka jest podstawowym składnikiem w tworzeniu organelli komórkowych zwanych peroksysomami (Genetics Home Reference, 2016).
W konsekwencji te mutacje genetyczne mogą prowadzić do niedostatecznego rozwoju biogenezy peroksysomów, a tym samym ich aktywności funkcjonalnej (Genetics Home Reference, 2016).
Diagnoza
Zespół Zellwegera można zdiagnozować w czasie ciąży lub w okresie poporodowym.
W przypadku diagnostyki prenatalnej USG kontroli ciąży może wykazywać różne nieprawidłowości zgodne z tą patologią, takie jak wewnątrzmaciczne opóźnienie wzrostu lub wady rozwojowe twarzoczaszki.
Niezbędne jest jednak przeprowadzenie analizy biochemicznej poprzez pobieranie próbek krwi i pobieranie próbek kosmków kosmówki w celu określenia obecności zaburzenia metabolicznego pochodzenia genetycznego.
Z drugiej strony, w przypadku diagnozy poporodowej, badanie fizykalne oferuje wystarczające wyniki kliniczne, aby potwierdzić jego obecność, chociaż wykonuje się różne testy w celu wykluczenia innych rodzajów patologii.
Niektóre testy laboratoryjne stosowane w diagnozie opierają się na badaniach histologicznych i biochemicznych lub testach neuroobrazowania (Cáceres-Marzal, Vaquerizo-Madryt, Girós, Ruiz i Roels, 2003).
Leczenie
Podobnie jak w przypadku innych rodzajów patologii genetycznych, nie zidentyfikowano jeszcze leku na zespół Zellwergera.
W tym przypadku interwencje medyczne są ukierunkowane na metody podtrzymywania życia i leczenie farmakologiczne.
Powikłania medyczne zwykle postępują wykładniczo, więc pogorszenie stanu klinicznego osób dotkniętych chorobą jest nieuniknione.
Większość osób dotkniętych zespołem Zellwegera zwykle nie przekracza 2 lat.