Zespół Wolfa-Hirschhorna: objawy, przyczyny, leczenie
Zespół Wolfa-Hirschhorna jest rzadką patologią genetyczną, której charakterystyka kliniczna wynika głównie z utraty materiału genetycznego (Hiszpańskie Stowarzyszenie Wolf-Hirschhorn Syndrome, 2012).
Na poziomie klinicznym patologia ta charakteryzuje się występowaniem zmian związanych z wadami rozwojowymi twarzy, epizodami drgawkowymi i znacznym uogólnionym opóźnieniem rozwoju (Aviña i Hernández, 2008).

Tak więc na określonym poziomie wiąże się z wieloma ważnymi powikłaniami medycznymi: urazami neurologicznymi, sercem, układem mięśniowo-szkieletowym, immunologicznym, wzrokowym, słuchowym, moczowo-płciowym itp. (Blanco-Lago, Malaga, García-Peñas, García-Rom, 2013).
Jeśli chodzi o etiologiczne pochodzenie zespołu Wolfa-Hirschhorna, jest ono związane z obecnością anomalii genetycznych na chromosomie 4 (Coppola, Chinthapalli, Hammond, Sander, Sisodiya, 2013).
Z drugiej strony, rozpoznanie zespołu Wolfa-Hirschhorna jest zwykle potwierdzane na etapie dzieciństwa, dzięki rozpoznaniu cech fizycznych i poznawczych. Jednak analiza genetyczna ma kluczowe znaczenie (Hiszpańskie Stowarzyszenie Wolf-Hirschhorn Syndrome, 2012).
Wreszcie interwencja terapeutyczna tej patologii opiera się zazwyczaj na rehabilitacji fizycznej, leczeniu logopedycznym, dostarczaniu leków przeciwpadaczkowych, adaptacjach dietetycznych lub interwencji neuropsychologicznej, wśród innych środków wsparcia (Medina, Rojas, Guevara, Cañizales i Jaimes, 2008). ).
Charakterystyka zespołu Wolfa-Hirschhorna
Zespół Wolfa jest patologią pochodzenia genetycznego, charakteryzującą się afektem mulsystemowym, zdefiniowanym przez obecność atypowych cech twarzy, uogólnione opóźnienie wzrostu, niepełnosprawność intelektualną i epizody drgawkowe (Genetics Home Reference, 2016).
Jednak przebieg kliniczny jest bardzo niejednorodny wśród osób dotkniętych chorobą, ze względu na genetyczny charakter tego zjawiska, będącego produktem delecji (National Organization for Rare Disroders, 2016).
Zatem przez delecję chromosomową rozumiemy utratę jednego lub więcej segmentów chromosomu (Chromosomal Mutations, 2016). W zależności od nasilenia tej anomalii i poziomu zaangażowania genetycznego, wśród dotkniętych chorobą mogą pojawić się cechy różnicowe (National Organization for Rare Disroders, 2016).
W tym sensie deficyt materiału genetycznego w zespole Wolfa jest związany z ważnymi komplikacjami medycznymi. Tak więc wiele osób dotkniętych chorobą umiera w fazie prenatalnej lub noworodkowej, jednak niektóre przypadki umiarkowanego nasilenia przekraczają pierwszy rok życia (Wolf Hirschhorn, 2013).
Chorobę tę opisali jednocześnie badacze Ulrich Wolf i Kart Hirschhon, równolegle w 1965 r. (Aviña i Hernández, 2008).
W pierwszych raportach klinicznych odniesiono się do zaburzenia charakteryzującego się obecnością mikrocefalii, z konfiguracją czaszki podobną do greckiego hełmu (Aviña i Hernández, 2008).
Jednak Zollino i jego grupa robocza opisali szczegółowo w 2001 r. Wszystkie cechy kliniczne zespołu Wolfa-Hirshhorna (Aviña i Hernández, 2008).
Do tej pory zidentyfikowano ponad 90 różnych przypadków w literaturze medycznej i eksperymentalnej, ogólnie związanych z płcią żeńską (Blanco-Lago, Malaga, García-Peñas, García-Rom, 2013).
Ponadto obecne definicje tej patologii obejmują zarówno identyfikację głównych lub kardynalnych objawów (atypowe fakty, opóźnienie wzrostu, opóźnienia rozwoju motorycznego i poznawczego oraz zmiany padaczkowe), jak i inne objawy medyczne (anomalie sercowe, czuciowe, moczowo-płciowe itp.). ) (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016).
Czy to częsta patologia?
Ogólnie rzecz biorąc, zespół Wolfa-Hirschhorna i jego charakterystyka kliniczna są uważane za rzadkie schorzenia genetyczne (Hiszpańskie Stowarzyszenie Wolf-Hirschhorn Syndrome, 2012).
Jednak pomimo niskiego rozpowszechnienia, niektóre badania statystyczne zdołały zidentyfikować dane związane z częstością występowania 1 przypadku na 50 000 urodzeń (Aviña i Hernández, 2008).
Jednak inni autorzy, tacy jak Blanco-Lago, Málaga, García-Peñas i García-Ron (2013), zwracają uwagę, że zespół Wolfa-Hirschhorna może osiągnąć częstość zbliżoną do 1 przypadku na 20 000 urodzeń.
Z drugiej strony, w odniesieniu do czynników socjodemograficznych związanych z zespołem Wolfa-Hirschhorna, stwierdzono większą częstość występowania w płci żeńskiej, szczególnie w stosunku 2: 1 w porównaniu z płcią męską (Medina i in., 2008),
Ponadto nie było możliwe zidentyfikowanie zróżnicowanej częstotliwości związanej z określonymi regionami geograficznymi lub określonymi grupami etnicznymi i / lub rasowymi (Medina i in., 2008).
Wreszcie, odnosząc się do czynników dziedzicznych, badania wykazały, że u ponad 80% osób dotkniętych tą patologią zawdzięcza losową mutację. Przypadki zespołu Wolfa-Hirschhorna dziedzicznego pochodzenia genetycznego są rzadkie (Medina i in., 2008).
Znaki i objawy
Jak wskazaliśmy wcześniej, objawy, które można zaobserwować u osób cierpiących na zespół Wolfa-Hirschhorna, mogą być bardzo zmienne, jednak zespół ten jest patologią zdefiniowaną przez kilka centralnych stanów medycznych (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016).
- Anomalie twarzy.
- Ogólne opóźnienie w rozwoju.
- Epizody konwulsyjne.
- Opóźnienie psychomotoryczne i poznawcze.
Anomalie twarzy
Charaktery czaszkowo-twarzowe są zwykle definiowane przez szeroką listę anomalii i zmian, które razem mają nietypowy wygląd twarzy, podobny do hełmów greckich wojowników (Wieckzorek, 2013).
Niektóre z najczęstszych ustaleń klinicznych w tym obszarze są związane z (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016, Krajowa Organizacja ds. Rzadkich Chorób, 2016, Genetics Home Reference, 2016):
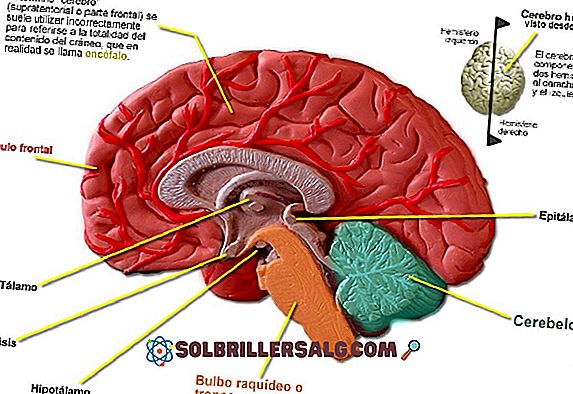
- Mikrocefalia : obwód czaszki zwykle nie rozwija się normalnie, więc całkowity rozmiar głowy jest zwykle niższy niż oczekiwany dla wieku chronologicznego osoby dotkniętej chorobą. Zasadniczo można zaobserwować różne asymetrie między różnymi strukturami tworzącymi obszar twarzoczaszki.
- Nosowa konfiguracja : nos ma zwykle nienormalnie duży rozmiar, którego górna część rozwija się płasko, z szerokim oddzieleniem obszaru między brwiami. W niektórych przypadkach nos przybiera nienormalny kształt, nazywany często nosem „dzioba papugi”.
- Konfiguracja twarzy : szczęka zwykle wydaje się słabo rozwinięta, będąc w stanie obserwować podbródek lub mały podbródek. Ponadto brwi zazwyczaj mają wysklepiony wygląd. Ponadto inne cechy patologiczne pojawiają się zwykle jako unaczynione plamy, między innymi wydaliny ze skóry.
- Wszczepienie pawilonu usznego : uszy są zwykle umieszczane w niższej pozycji niż zwykle. Ponadto można zaobserwować niedorozwój uszu, wyglądający na mniejszy i bardziej widoczny niż zwykle.
- Konfiguracja oka : oczy zwykle wydają się być szeroko oddzielone i mają znaczącą symetrię, będąc mniejszym z gałek ocznych. Ponadto możemy zidentyfikować zez, zmiany w strukturze i zabarwieniu tęczówki, opadające powieki lub niedrożność przewodów łzowych.
- Zmiany w jamie ustnej : w przypadku konfiguracji policzkowej najczęstszą jest identyfikacja nieprawidłowo niewielkiego filtru wargowego, rozszczepu wargi, opóźnionego wylęgu zębów, rozszczepu podniebienia, między innymi.
Uogólnione opóźnienie w rozwoju
W zespole Wolfa-Hirschhorna można zidentyfikować ogólne opóźnienie wzrostu i rozwoju, zarówno w stadiach prenatalnych, jak i postnatalnych i dziecięcych (Aviña i Hernández, 2008).
W tym sensie dzieci cierpiące na tę patologię mają tendencję do nadmiernego wzrostu, dlatego zazwyczaj mają mniejszą wagę i wzrost niż oczekiwano ze względu na płeć i wiek chronologiczny (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016 ).
Ten typ cech zwykle nie wiąże się z trudnościami w karmieniu lub niedoborami w przyjmowaniu kalorii, jednak zarówno zmiany genetyczne, jak i rozwój innych rodzajów patologii, takich jak zmiany w sercu, mogą przyczynić się do pogorszenia tego stanu zdrowia (Asociación Española zespołu Wolfa-Hirschhorna, 2016).
Ponadto uogólnione opóźnienie wzrostu jest zwykle związane z różnymi nieprawidłowościami układu mięśniowo-szkieletowego:
- Niedorozwój mięśniowy : struktura mięśniowa zwykle nie rozwija się całkowicie, dlatego bardzo często obserwuje się nienormalnie zmniejszone napięcie mięśniowe.
- Skolioza i kifoza : struktura kości kręgosłupa może być utworzona wadliwie, przedstawiając odchyloną pozycję lub z nieprawidłową krzywizną.
- Clinodactyly : struktura kości palców również zwykle rozwija się nieprawidłowo, dlatego można zaobserwować odchylenia w palcach. Ponadto ma tendencję do
zidentyfikować zmiany w konfiguracji odcisków palców.
- Nienormalnie cienkie kończyny : niska waga jest szczególnie widoczna w ramionach i nogach.
Epizody konwulsyjne
Napady padaczkowe są jednym z najczęstszych i najpoważniejszych objawów zespołu Wolfa-Hirschhorna (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016).
W tym sensie ataki są definiowane jako proces patologiczny wynikający z niezwykłej aktywności neuronalnej, która jest zmieniona, powodując pobudzenie ruchowe, skurcze mięśni lub okresy nietypowego zachowania i odczuć i czasami może powodować utratę przytomności (Mayo Clinic ., 2015).
W przypadku syndromu zespołu Wolfa-Hirschhorna najczęstszymi kryzysami są toniczno-kloniczne (Hiszpańskie Stowarzyszenie Syndromu Wolfa-Hirschhorna, 2016).
Zatem epizody drgawkowe charakteryzują się rozwojem napięcia mięśniowego, z tendencją do uogólnionej sztywności, zwłaszcza w nogach i ramionach, po której następują powtarzające się i niekontrolowane skurcze mięśni. Na poziomie wizualnym można je zaobserwować jako drżenie ciała (National Institute of Neuropathic Disorders and Stroke, 2015).
Ponadto nasilenie tego zdarzenia zależy od jego wpływu na tkankę mózgową. Nieprawidłowa i / lub patologiczna aktywność neuronalna może wpływać na dużą część struktury mózgu lokalnie lub uogólnioną, co może mieć istotne konsekwencje i następstwa neurologiczne (Narodowy Instytut Zaburzeń Neurologicznych i Udar mózgu, 2016).
Opóźnienie psychomotoryczne i poznawcze
W przypadku sfery kognitywnej ponad 75% osób dotkniętych zespołem Wolfa-Hirschhorna przedstawia pewien rodzaj niepełnosprawności intelektualnej (Medina, Rojas, Guevara, Cañizales i Jaimes, 2008).
Ogólnie rzecz biorąc, afektacja intelektualna jest zazwyczaj poważna, zazwyczaj nie rozwijają zdolności językowych, dlatego w dużej części przypadków komunikacja ogranicza się do emisji niektórych dźwięków (Medina, Rojas, Guevara, Cañizales i Jaimes, 2008).
Ponadto, w przypadku nabywania kontroli postawy, stania, chodzenia itp., Wszystkie te są znacznie opóźnione, głównie z powodu nieprawidłowości układu mięśniowo-szkieletowego.
Kurs kliniczny
W większości przypadków objawy rozwijają się stopniowo, tak że w rozwoju tej patologii można wyróżnić kilka etapów (Wieczorek, 2003):
- Pierwszy rok życia : w najwcześniejszych fazach najbardziej charakterystyczne objawy są związane z małą masą ciała i anomaliami twarzoczaszki. W wielu przypadkach, około 35%, osoby dotknięte chorobą umierają z powodu równoległej obecności wrodzonych wad serca.
- Stopień rozwojowy dziecka : oprócz opóźnienia w rozwoju fizycznym, zwłaszcza wady układu mięśniowo-szkieletowego są szczególnie widoczne. Wraz z tymi odkryciami medycznymi pojawiają się nawracające napady. Generalnie niewiele osób dotkniętych chorobą jest w stanie chodzić lub opanować język.
- Późne dzieciństwo i okres dojrzewania : na tym etapie najważniejsze są cechy związane z rozwojem i funkcjonowaniem intelektualnym, jednak typowe rysy twarzy stają się oczywiste.
Przyczyny
Jak zauważyliśmy w początkowym opisie zespołu zespołu Wolfa-Hirschhorna, zaburzenie to spowodowane jest delecją genetyczną zlokalizowaną na chromosomie 4 (Genectis Home Reference, 2016).
Chociaż objętość utraty materiału genetycznego może się znacznie różnić u osób dotkniętych chorobą, im bardziej poważna i znacząca, tym ostrzejsza będzie symptomatologia związana z tą chorobą (Genectis Home Reference, 2016).
Chociaż nie wszystkie zaangażowane geny są dokładnie znane, różne badania powiązały brak genów WHSC1, LEMT1 i MSX1 z przebiegiem klinicznym zespołu Wolfa-Hirschhorna (Genectis Home Reference, 2016).
Diagnoza
Możliwe jest postawienie diagnozy zespołu Wolfa-Hirschhorna przed urodzeniem (National Organization for Rare Disorders, 2016).
Ultradźwięki kontroli ciążowej umożliwiają identyfikację zaburzeń wzrostu wewnątrzmacicznego i innych rodzajów wad fizycznych (National Organization for Rare Disorders, 2016).
Jednak konieczne jest przeprowadzenie badania genetycznego w celu potwierdzenia stanu, poprzez przed- lub poporodową analizę komórkową (National Organization for Rare Disorders, 2016).
Leczenie
Obecnie nie ma lekarstwa na zespół Wolfa-Hirschhorna ani standardowe podejście terapeutyczne, więc leczenie jest zaprojektowane specjalnie według indywidualnych cech i przebiegu klinicznego patologii (Wolf Hirschhorn, 2013).
Interwencja medyczna koncentruje się zatem zazwyczaj na leczeniu napadów poprzez podawanie leków przeciwpadaczkowych, suplementacji żywieniowej, chirurgicznej korekcji wad fizycznych, rehabilitacji poznawczej i edukacji specjalnej (Wolf Hirschhorn, 2013).