Zespół Ushera: objawy, przyczyny, leczenie
Zespół Ushera składa się z grupy zaburzeń wrodzonego pochodzenia dziedzicznego charakteryzujących się zmianami neurosensorycznymi (Nàjera, Baneyto i Millán, 2005).
Na poziomie klinicznym patologię tę określa obecność obustronnej głuchoty, barwnikowego zwyrodnienia siatkówki i różnych zmian przedsionkowych (Nàjera, Baneyto i Millán, 2005).

W zależności od nasilenia i dotkniętych obszarów, zespół Ushera jest zwykle podzielony na trzy formy kliniczne: zespół Ushera I (USH1), zespół Ushera II (USH2) i zespół Ushera III (USH3) (Jaijo, Aller, Beneyto, Nájera i Millán, 2005).
Etiologiczna przyczyna tego zespołu jest związana z autosomalnym recesywnym wzorem zdefiniowanym przez szeroką heterogeniczność genetyczną (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011).
Zidentyfikowano ponad 8 różnych genów związanych z pojawieniem się zespołu Ushera. Są one odpowiedzialne za każdy z podtypów klinicznych (López, Gelvez i Tamayo, 2011).
Rozpoznanie tej choroby wymaga zastosowania różnych analiz okulistycznych i audiologicznych. Ponadto zazwyczaj przeprowadza się badanie genetyczne w celu analizy specyficznych mutacji (Sabaté Cintas, 2009).
Nie ma leczniczego podejścia terapeutycznego do tego zaburzenia. Najczęściej stosuje się metody adaptacji fizycznej, rehabilitacji, szkolenia w zakresie orientacji / mobilności i kształcenia specjalnego (Sabaté Cintas, 2009).
Ponadto rokowanie medyczne osób dotkniętych chorobą zwykle charakteryzuje się postępującym rozwojem psychologicznych i / lub neurologicznych zmian psychologicznych, które znacząco pogarszają jakość ich życia (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011 ).
Charakterystyka zespołu Ushsera
Zespół Ushera (SU) jest jedną z najczęstszych przyczyn ślepoty i głuchoty pochodzenia genetycznego (American Academy of Ophthalmology, 2013).
Jest to choroba charakteryzująca się obrazem klinicznym pogorszenia słuchu o charakterze sensorycznym, utratą ostrości wzroku i anomaliami przedsionkowymi (American Academy of Ophthalmology, 2013).
Kurs kliniczny jest powiązany z (Nàjera, Baneyto i Millán, 2005):
- Urazy i anomalie ucha wewnętrznego (zaburzenia słuchu i równowagi).
- Zwyrodnienie barwnikowe siatkówki (zaburzenia widzenia)
Zaburzenie to jest szczególnie definiowane przez jego zmienność kliniczną i genetyczną. Badania kliniczne zwykle używają terminu Zespół Ushera jako grupy zaburzeń (USH1, USH2 i USH3) (Genetics Home Reference, 2016).
Jest to choroba o dużym zainteresowaniu medycznym i psychologicznym ze względu na stopień izolacji sensorycznej i społecznej, którą dotknęły osoby dotknięte chorobą (Jaijo i in., 2005).
Pierwsze opisy kliniczne tego zaburzenia są spowodowane przez Von Graefe i Libreich, którzy zidentyfikowali znaczące medyczne powiązanie między głuchotą a barwnikowym zwyrodnieniem siatkówki (Braga Norte, Cortez Juares, Nardi, Dell'Aringa i Kobari, 2007).
Jego dziedziczny charakter został zidentyfikowany w 1914 r. Dzięki badaniom brytyjskiego okulisty Ushera, od którego otrzymał nazwisko (Cleveland Clinic, 2016).
Jednak Bell (1933) był jednym z pionierów w identyfikacji wielkiej klinicznej różnorodności, która definiuje ten zespół (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011).
Statystyki
Większość badań klinicznych, epidemiologicznych i / lub eksperymentalnych uważa, że zespół Ushera jest częścią rzadkich lub rzadkich chorób (Wallber, 2009).
Jednak zespół Ushera jest najczęstszą przyczyną ślepoty u ludzi (Wallber, 2009).
Pochodzenie charakterystyki klinicznej 6% wrodzonych osób głuchych i 18% osób cierpiących na barwnikowe zwyrodnienie siatkówki wynika ze stanu zespołu Ushera (López, Gelvez i Tamayo, 2011).
Ogólną częstość występowania tego zespołu szacuje się na około 3-4 przypadki na 100 000 osób w populacji ogólnej, jeśli jest to specyficzny związek z płcią, rasą lub pochodzeniem geograficznym (Sabaté Cintas, 2009).
Jednak inni autorzy, tacy jak López, Gelvez i Tamayo (2011), umieszczają częstość występowania na 3, 5-6, 2 przypadków na 100 000 osób.
W przypadku Hiszpanii wskaźniki rozpowszechnienia mogą osiągnąć 4, 2 przypadków na 100 000 mieszkańców, zakładając, że na całym terytorium dotknie ich około 1600 osób (Jaijo, Aller, Beneyto, Nájera i Millán, 2005).
W Stanach Zjednoczonych znajduje się w około 5 przypadkach na 100 000 mieszkańców; w regionach skandynawskich w 3 na 100 000 iw Kolumbii w liczbie bliskiej 3, 2 przypadków na 100 000 osób (López, Gelvez i Tamayo, 2011).
Wreszcie, jeśli chodzi o dystrybucję przypadków według podtypów, możemy wskazać następujące dane (Genetics Home Reference, 2016):
- Typ I i II jako najczęstsze formy zespołu Ushera.
- Typ III, najmniej wspólny, stanowiący 2% wszystkich przypadków.
Znaki i objawy
Charakterystyka kliniczna zespołu Ushera jest zasadniczo związana z głuchotą czuciowo-nerwową, utratą ostrości wzroku i zmianą układu przedsionkowego.
Głuchota odbiorczo-nerwowa
Poziom ostrości słuchowej może się znacznie różnić u osób dotkniętych chorobą i w zależności od podtypu cierpiącego na zespół Ushera (Sabaté Cintas, 2009).
Osoby mogą cierpieć na całkowitą wrodzoną głuchotę, umiarkowane problemy ze słuchem lub prawidłową lub sprawną ostrość wzroku (Sabaté Cintas, 2009).
Wszystkie problemy związane z obszarem słuchowym mają swój początek w obecności zmiany neurosensorycznej. Dlatego najczęściej obserwuje się rodzaj głuchoty odbiorczej lub niedosłuchu (Genetics Home Reference, 2016).
Ta patologia odnosi się do obecności wrodzonych zmian w uchu wewnętrznym i zmiennej zmiany włókien i zakończeń nerwowych związanych z nerwem słuchowym (Cochlear, 2016).
Strata ostrości wzroku
Zaburzenia widzenia są zwykle podstawową zmianą kliniczną zespołu Ushera (American Academy of Ophthalmology, 2016).
Osoby dotknięte chorobą prezentują przebieg charakteryzujący się postępującym zmniejszeniem ostrości wzroku, zdefiniowany następującym wzorem (Genetics Home References, 2016):
- Utrata widzenia w nocy.
- Utrata widzenia bocznego.
- Pojawienie się martwych punktów.
- Rozwój zmętnienia soczewki (zaćmy).
Wszystkie te anomalie okulistyczne mają swoje źródło w prezentacji barwnikowego zwyrodnienia siatkówki (RP).
Zapalenie siatkówki barwnikowe jest stanem medycznym, który odnosi się do postępującego rozwoju zmian chorobowych w komórkach ocznych wrażliwych na światło (American Academy of Ohthomology, 2016).
Komórki te, zwane stożkami i prętami, znajdują się w siatkówce i są zdolne do przekształcania bodźców świetlnych w sygnały elektryczne interpretowane na poziomie mózgowym (American Academy of Ohtalomology, 2016).
Częstość występowania różnych czynników, takich jak nieprawidłowości genetyczne, może prowadzić do śmierci tych komórek (American Academy of Ohtalomology, 2016).
Początkowo wpływa na laski, głównie odpowiedzialne za widzenie nocne i obwodowe. Następnie następuje pogorszenie stożków, odpowiedzialnych za widzenie centralne i postrzeganie kolorów (American Academy of Ohtalomology, 2016).
Zmiana systemu przedsionkowego
Wrodzone anomalie obecne w uchu wewnętrznym mogą również powodować pewne znaczące zmiany w układzie przedsionkowym (Nàjera, Baneyto i Millán, 2005).
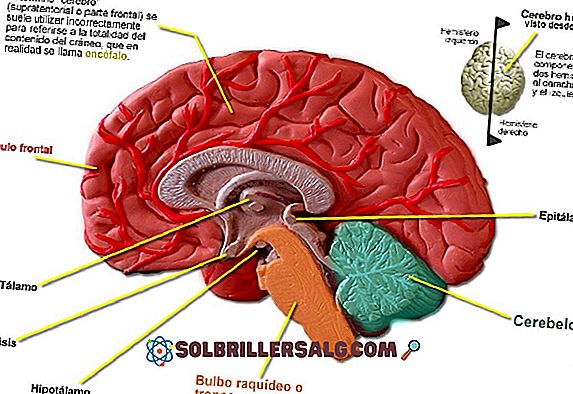
Układ przedsionkowy składa się z różnych struktur, które odgrywają zasadniczą rolę w równowadze i skutecznym utrzymaniu postawy ciała.
System ten grupuje kilka elementów obwodowych (przedsionkowe zakończenia nerwowe i ucho wewnętrzne) i inne o charakterze centralnym na poziomie mózgu i kręgosłupa.
W zespole Ushera zaangażowanie niektórych z tych składników spowoduje różne objawy zasadniczo związane z równowagą (Genetics Home Reference, 2016).
W konsekwencji często obserwuje się problemy z orientacją, częstą utratą równowagi, nabyciem pozycji siedzącej i późnej, między innymi (Genetics Home Reference, 2016).
Jakie są różne podtypy?
Zespół Ushera można podzielić na kilka podtypów w zależności od wieku, w którym pojawiają się pierwsze objawy, charakterystyki klinicznej i ciężkości stanu medycznego (Jaijo, Aller, Beneyto, Nájera i Millán, 2005).
Zespół Ushera Typ I
Pierwszy podtyp syndromu Ushera można zidentyfikować od urodzenia, chociaż niektóre specyficzne cechy są postępowe (Sabaté Cintas, 2009):
Anomalie słuchowe charakteryzują się głęboką głuchotą wrodzoną, to znaczy od urodzenia. Ponadto nie ma możliwości zastosowania określonych adaptacji, takich jak aparaty słuchowe, aby poprawić tę pojemność.
Zmiany wizualne wydają się być podstępne. Pierwsze problemy ze wzrokiem pojawiają się około 10 lat i wraz z wiekiem mogą rozwinąć się w ślepotę.
Możliwe jest również zidentyfikowanie nieprawidłowości związanych z układem przedsionkowym. Te zasadniczo poprzez poważne problemy równowagi.
Syndrom Ushera typu II
Podtyp II zespołu Ushera stanowi późniejszy debiut. Typowe epoki pojawienia się pierwszych objawów są zwykle zlokalizowane na etapie dorastania (Sabaté Cintas, 2009):
Zmiany słuchowe mają zwykle mniej poważny charakter. Chociaż możliwy jest rozwój umiarkowanych deficytów słuchu, możliwe jest użycie aparatów słuchowych w celu poprawy ich wydajności.
Ponadto obecność szczątkowego słuchu pozwala im na używanie języka ustnego jako podstawowego środka komunikacji.
Deficyty wzrokowe są zwykle związane z postępującym rozwojem barwnikowego zapalenia siatkówki, podczas gdy równowaga nie jest znacząco zmieniona.
Zespół Ushera typu III
Trzeci i ostatni podtyp zespołu Ushera ma typową prezentację w wieku dorosłym. Chociaż niektóre cechy kliniczne mogą wystąpić wcześniej (Sabaté Cintas, 2009):
Ostrość słuchu charakteryzuje się normalnym lub standardowym początkiem, który musi być zmniejszony w wieku dorosłym, prowadząc do głuchoty.
Anomalie wzrokowe są definiowane przez prezentację barwnikowego zapalenia siatkówki u nastolatków i rozwój ślepoty podczas pośrednich etapów stadium dorosłego.
W końcu wpływa to również na układ przedsionkowy, co prowadzi do rozwoju ważnych problemów z koordynacją i równowagą.
Przyczyny
Jak wskazaliśmy w początkowym opisie, zespół Ushera ma autosomalne recesywne dziedziczne pochodzenie (López, Gelvez i Tamayo, 2011).
Zmiany genetyczne są zasadniczo definiowane przez heterogeniczność, ponieważ różne anomalie odpowiadają każdemu z różnych podtypów (López, Gelvez i Tamayo, 2011).
Możliwe było zidentyfikowanie ponad 12 lokalizacji różnych zmian genetycznych, którym towarzyszyło ponad 8 specyficznych mutacji: MYO7A, USH3, USH1C, VLGR1, CDH23, SANS, CLRN1, OCDH15 (Nàjera, Baneyto i Millán, 2005).
Większość przypadków typu I wiąże się z mutacjami genu MYO7A i CDH12. Podczas gdy typ II jest bardziej związany ze specyficznymi mutacjami genu USH2A. Wreszcie, typ III jest spowodowany mutacjami w genie CLRN1 (Genetics Home Reference, 2016).
Diagnoza
Charakterystyka kliniczna zespołu Ushera opiera się na rozpoznaniu systemu słuchowego, oftalmologicznego i przedsionkowego (American Academy of Ophtalmology, 2016).
Dlatego istotne jest, aby ocenić pojemność słuchową, ostrość widzenia i obecność możliwych zmian równowagi i koordynacji ciała (American Academy of Ophtalmology, 2016).
- Badanie słuchowe : audiometria, emisje otoakustyczne, potencjały wywołane ślimakiem i otoskopia (Sabaté Cintas, 2009).
- Badanie okulistyczne : dno, kampimetria, elektroretinogram, elektrookulogram i elektronystagmogram.
- Badanie przedsionkowe : chociaż niektóre z poprzednich testów mogą zidentyfikować pewne zmiany w układzie przedsionkowym, najczęściej wykonuje się test równowagi.
Oprócz opisanych powyżej podejść niezwykle ważne jest przeprowadzenie badania genetycznego ze względu na dziedziczną naturę tej patologii.
Podstawowym celem tego typu testów jest zidentyfikowanie specyficznej mutacji genetycznej, która powoduje powstanie podtypu klinicznego, na który cierpi pacjent, i zidentyfikowanie ich wzorca dziedziczności.
Leczenie
Nie ma lekarstwa ani podejścia terapeutycznego zaprojektowanego specjalnie dla zespołu Ushera (Sabaté Cintas, 2009).
Różni specjaliści i instytucje, takie jak American Academy of Ophthalmoogy (2016), podkreślają, że najlepszym podejściem sanitarnym jest identyfikacja i wczesna diagnoza.
Klasyczne terapie obejmują:
- Urządzenia do kompensacji słuchowej, takie jak implant ślimakowy.
- Wizualne urządzenia kompensacyjne, takie jak soczewki lub adaptacje.
- Terapia witaminami oparta na podawaniu witaminy A do kontroli barwnikowego zapalenia siatkówki.
- Rehabilitacja fizyczna w celu poprawy równowagi ciała i problemów z koordynacją.
- Terapia komunikacyjna do generowania alternatywnych form komunikacji.
Ponadto trwają badania nad terapiami alternatywnymi nowej generacji, wszystkie związane z wymianą genetyczną.