Zespół Lennoxa-Gastauta: objawy, przyczyny, leczenie
Zespół Lennoxa-Gastauta ( SLG ) jest rodzajem padaczki o ogromnym nasileniu związanym z wiekiem. Charakteryzuje się odpornością na leczenie farmakologiczne i różnorodnością powodowanych przez nie niepełnosprawności (Valdivia Álvarez i Marreno Martínez, 2012).
Jest to zaburzenie, które zwykle objawia się w dzieciństwie, z początkiem między 3 a 5 rokiem życia. Może cierpieć nawet 6% wszystkich dzieci z padaczką (David, García i Meneses, 2014).

Na poziomie klinicznym zespół ten jest definiowany przez występowanie napadów tonicznych, toniczno-klonicznych lub mioklonicznych, którym towarzyszy zmienny stopień niepełnosprawności intelektualnej (Rey, Encabo, Pizarro, San Martín i López-Timoneda, 2015).
Etiologiczne pochodzenie zespołu Lennoxa-Gastauta może być związane z wieloma czynnikami, w tym między innymi: zmianami genetycznymi, patologiami nerwowo-skórnymi, wypadkami mózgowo-naczyniowymi, procesami zakaźnymi na poziomie mózgu lub urazami czaszkowo-mózgowymi (David, García i Meneses, 2014).
Biorąc pod uwagę podejrzenie patologii padaczki, diagnoza tego zespołu opiera się głównie na analizie napadów i nagrań elektroencefalograficznych (EGG) (Campos Castelló, 2007).
Obecnie nie zidentyfikowano skutecznego leczenia zespołu Lennoxa-Gastauta (Fernández, Serrano, Solarte, Cornejo, 2015).
Zazwyczaj stosuje się pewne podejścia terapeutyczne, takie jak podawanie leków przeciwpadaczkowych nowej generacji, przepisywanie diety ketogenicznej, stymulacja nerwu błędnego, paliatywna interwencja medyczna lub operacja (Kim, Kim, Lee, Heo, Kim i Kang, 2015).
Charakterystyka zespołu Lennoxa-Gastauta
Zespół Lennoxa-Gastauta ( SLG ) jest formą padaczki dziecięcej. Przedstawia poważny przebieg kliniczny zdefiniowany przez rozwój wielu napadów i zmiennej niepełnosprawności intelektualnej (Narodowe Centrum Zaawansowanych Nauk Translacyjnych, 2016).
Padaczkę można zdefiniować jako zaburzenie neurologiczne charakteryzujące się występowaniem kolejnych lub nawracających epizodów zwanych napadami padaczkowymi lub napadami padaczkowymi (Fernández-Suárez i in., 2015).
Jest to rodzaj choroby o ogromnej częstości występowania na całym świecie. Światowa Organizacja Zdrowia (2016) zgłosiła ponad 50 milionów przypadków na całym świecie.
Pochodzi z obecności funkcjonalnych lub strukturalnych zmian w układzie nerwowym (SN). Ponadto może dotyczyć każdego, niezależnie od wieku i płci.
U dzieci padaczka jest częstym zaburzeniem z heterogenicznymi objawami klinicznymi i jest bardzo związana z rozwojem biologicznym i wiekiem (López, Varela i Marca, 2013).
Chociaż można wyróżnić wiele różnych rodzajów padaczki dziecięcej, wszystkie z nich zwykle stanowią wspólny czynnik: predyspozycje do cierpienia z powodu kryzysu z dużą częstotliwością (López, Varela i Marca, 2013).
Mają szeroko heterogeniczne prognozy medyczne, różne powiązane patologie i wysoce zróżnicowaną odpowiedź na podejścia terapeutyczne (López, Varela i Marca, 2013).
W tym sensie istnieje zmniejszona grupa zespołów i form padaczkowych, które stanowią wzorzec oporny lub oporne na leki przeciwpadaczkowe (López, Varela i Marca, 2013).
Jedna z tych grup chorób odpowiada encefalopatiom padaczkowym, w których zespół Lennoxa-Gastauta jest zwykle klasyfikowany (López, Varela i Marca, 2013).
Termin encefalopatia padaczkowa odnosi się do szerokiego zestawu poważnych stanów konwulsyjnych, które zwykle zaczynają przebieg kliniczny we wczesnych etapach życia (pierwsze dni życia lub wczesne dzieciństwo) (Aviña Fierro i Hernández Aviña, 2007).
Zespoły te mają tendencję do postępu w kierunku postaci nieleczonych padaczek, z poważnym rozwojem symptomatologicznym. W większości przypadków mają fatalny skutek (Aviña Fierro i Hernández Aviña, 2007).
Pierwsze opisy tego zespołu w 1950 r. Odpowiadają badaczom Lennoxa i Davisa (Valdivia Álvarez i Marreno Martínez, 2012).
Dzięki rozwojowi klinicznej elektroencefalografii (EGG) autorzy zdołali ustalić związek między aktywnością neuronalną a objawami klinicznymi badanych pacjentów (Oller-Durela, 1972).
Po latach Gastaut (1966) i inni badacze uzupełnili kliniczny opis tej patologii (Valdivia Álvarez i Marreno Martínez, 2012).
Gastautowi udało się opisać przebieg kliniczny serii 100 różnych przypadków. Jednak to Niedermeyer (1969) ostatecznie wprowadził nazwę tej patologii w dziedzinie medycyny i eksperymentów (David, García i Meneses, 2014)
Początkowo Międzynarodowa Klasyfikacja Padaczki uważała zespół Lennoxa-Gastauta za rodzaj uogólnionej padaczki o charakterze kryptogennym lub bezobjawowym (Herranz, Casas-Fernández, Campistol, Campos-Castelló, Rufo-Campos, Torres, Falcón i de Rosendo, 2010).
Najbardziej aktualne definicje, takie jak proponowane przez Centralną Ligę Padaczki, odnoszą się do zespołu Lennoxa-Gastauta jako formy pierwotnej, uogólnionej padaczki i katastrofalnej lub bardzo poważnej ekspresji klinicznej (Herranz i in., 2010).
Statystyki
Zespół Lennoxa-Gastauta uważany jest za jeden z najpoważniejszych typów lub form padaczki u dzieci (Valdivia Álvarez i Marreno Martínez, 2012).
Zaburzenie to stanowi zwykle przyczynę około 2-5% całkowitej liczby padaczek u dzieci lub dzieci (Epilepsy Foundation, 2016).
Chociaż może rozwinąć się w każdej grupie wiekowej, typowy początek wynosi od 3 do 5 lat (David, García i Meneses, 2014).
W Stanach Zjednoczonych badania epidemiologiczne oceniają częstość występowania zespołu Lennoxa-Gastauta u około 14 500–18 500 dzieci w wieku poniżej 18 lat (Fundacja Lennox-Gastaut, 2016).
Jest to zazwyczaj częstsza choroba u dzieci (0, 1 na 1000 osób) niż u dziewcząt (0, 02 na 1000 osób) (Cherian, 2016).
Jeśli chodzi o charakterystykę kliniczną, około 90% osób z rozpoznaniem zespołu Lennoxa-Gastauta cierpi z powodu pewnego rodzaju niepełnosprawności lub opóźnienia intelektualnego od początku choroby (Valdivia Álvarez i Marreno Martínez, 2012).
Ponadto ponad 80% przewlekle cierpi na różne rodzaje napadów (Valdivia Álvarez i Marreno Martínez, 2012).
Analiza przyczyn zespołu Lennoxa-Gastauta pokazuje, że około 30% wszystkich przypadków ma zidentyfikowaną etiologię, bez wcześniejszych przypadków neurologicznych. Podczas gdy 60% ma powiązane zaburzenia neurologiczne (Rey, Encabo, Pizarro, San Martín i López-Timoneda, 2015).
Znaki i objawy
Zespół Lennoxa-Gastauta charakteryzuje się trzema podstawowymi odkryciami: wzorzec fal elektroencefalograficznych, drgawki i zmienna niepełnosprawność intelektualna (Fernández Echávez, Serrano Tabares, Solarte Mila i Cornejo Ochoa, 2015).
Wzór elektroencefalograficzny
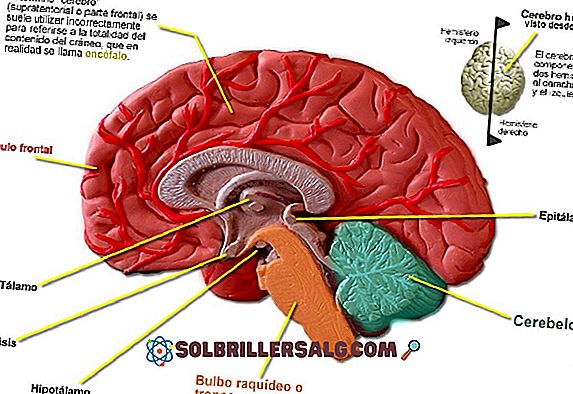
Oprócz aktywności biochemicznej, wzorce elektryczne mają podstawowe znaczenie dla funkcjonowania mózgu.
Aktywność elektryczna jest jedną z najszybszych i najskuteczniejszych form komunikacji między składnikami neuronalnymi naszego układu nerwowego.
Na poziomie globalnym możemy zidentyfikować grupy neuronów, które mają tendencję do aktywacji w skoordynowany i synchroniczny sposób w stanie spoczynku lub przed wykonaniem pewnego rodzaju określonego zadania.
Koordynacja ta jest zwykle opisywana jako wzory fal elektrycznych o mniejszej lub większej amplitudzie, w zależności od wykonywanej przez nas aktywności lub obszarów mózgu.
Istnieją różne rodzaje fal mózgowych: Delta, Theta, Alpha, Beta, klasyfikowane wokół ich częstotliwości, wolne lub szybkie.
W przypadku zespołu Lennoxa-Gastauta aktywność mózgu ma tendencję do dezorganizacji i asynchroniczności, powodując trwałe wzorce powolnych fal, typowe dla faz snu.
Autorzy tacy jak Díaz Negrillo, Martín del Valle i González Salaices, Prieto Jurczynska i Carneado Ruiz (2011) definiują te wzorce jako interciticową aktywność elektroencefalograficzną uogólnionych powolnych fal o częstotliwości od 1, 5 do 2, 5 hz podczas fazy czuwania i szybkiego działania rytmiczny podczas fazy snu.
Napady drgawkowe
Nieprawidłowa aktywność elektryczna neuronów w zespole Lennoxa-Gastauta prowadzi do rozwoju napadów, typowych dla padaczkowych form medycznych.
Atak lub napad charakteryzuje się wywołaniem wzorca nieprawidłowego zachowania w ograniczonym okresie czasu: mimowolne skurcze mięśni, postrzeganie niezwykłych odczuć, utrata przytomności itp. (Mayo Clinic., 2015).
W zależności od charakterystyki klinicznej i prezentacji napadów padaczkowych możemy odróżnić różne typy.
W przypadku zespołu Lennoxa-Gastauta najczęściej występują napady toniczne, toniczno-kloniczne lub miokloniczne (Rey, Encabo, Pizarro, San Martín i López-Timoneda, 2015).
Wszystkie te zazwyczaj przedstawiają ogólną prezentację. Ten wzorzec początku charakteryzuje się globalnym zaangażowaniem struktur mózgu (Mayo Clinic., 2015).
Nieprawidłowa aktywność neuronalna musi być generowana w określonym obszarze lub obszarze i rozwijać się w kierunku pozostałych obszarów mózgu (Mayo Clinic., 2015).
W oparciu o klasyfikację andaluzyjskiego stowarzyszenia padaczki (2016), opiszemy niektóre z najważniejszych cech tego typu kryzysu:
Kryzys toniczny
Napady toniczne lub napady padaczkowe są definiowane przez nagły rozwój wysokiego napięcia mięśniowego, to znaczy znaczną sztywność ciała.
Ta zmiana mięśni zwykle powoduje utratę stabilności ciała, a zatem upadek na ziemię.
Rzadko zdarza się, że występują w izolacji, ponieważ zwykle towarzyszy im faza kloniczna.
Kryzys toniczno-kloniczny
W tym przypadku kryzys zwykle zaczyna się od ogólnej sztywności całego ciała (epizod toniczny), która prowadzi do rozwoju mimowolnych i niekontrolowanych ruchów mięśni (epizody kloniczne).
Ogólnie ruchy są rytmiczne i dotyczą kończyn, głowy lub tułowia ciała.
Mogą one powodować pewne komplikacje: ukąszenia językowe, posiniaczone usta, utratę moczu lub urazy wtórne do nagłych upadków.
To przejściowe kryzysy. Osoba dotknięta chorobą powraca stopniowo po kilku minutach.
Napady toniczno-kloniczne są uważane za najpoważniejsze i uciążliwe z powodu ich objawów.
Kryzys miokloniczny
Ten rodzaj kryzysu jest definiowany przez nagłe gwałtowne szarpnięcia mięśni.
Może wpływać na całą strukturę ciała lub niektóre specyficzne regiony, takie jak kończyny górne lub dolne.
W większości przypadków powodują utratę stabilności ciała, spadając na ziemię lub spadające przedmioty.
Mają ograniczony czas trwania, około kilku sekund. Są uważane za łagodniejsze niż poprzednie formy.
Kryzys nieobecności
Chociaż są rzadsze, mogą pojawić się również nietypowe kryzysy nieobecności (Genetics Home Reference, 2016).
Ten typ zdarzenia medycznego charakteryzuje się częściową lub całkowitą utratą świadomości i połączeniem ze środowiskiem (Genetics Home Reference, 2016).
Wielu pacjentów cierpi na nagłą utratę napięcia mięśniowego równolegle, więc kryzysy związane z nieobecnością są zwykle związane z upadkami i różnego rodzaju wypadkami traumatycznymi (Genetics Home Reference, 2016).
Niepełnosprawność intelektualna
Nieprawidłowa lub patologiczna aktywność elektryczna towarzysząca zespołowi Lennoxa-Gastauta powoduje postępujące pogorszenie struktur nerwowych w mózgu.
W konsekwencji u wielu osób dotkniętych chorobą można zidentyfikować różne zmiany poznawcze, którym towarzyszy zmienna niepełnosprawność intelektualna .
Badania kliniczne wskazują, że opóźnienie rozwoju neurologicznego stanowi jedno z ustaleń klinicznych obecnych od momentu rozpoznania (Fundacja Lennox-Gasteau, 2016).
Jedną z najbardziej znaczących cech jest identyfikacja wyraźnego opóźnienia psychomotorycznego . Zwykle jest definiowany przez obecność (Asociación Andaluza de Epilepsia, 2016):
- Niestabilność ciała.
- Hiperkineza
U wielu dotkniętych chorobą rozpoznanie zespołu Lennoxa-Gasteau zwykle obejmuje inne równoległe rozpoznania:
- Zaburzenia neuropsychiatryczne.
- Uogólnione zaburzenia rozwojowe.
Zwykle występują różne związane z nimi anomalie behawioralne (David, García i Meneses, 2014):
- Agresywne zachowanie
- Autystyczne tendencje
- Zmiany osobowości.
- Nadpobudliwość
Osoby dotknięte zespołem Lennoxa-Gastauta spędzą całe życie cierpiąc na zaburzenia poznawcze i zmiany behawioralne i społeczne (Fundacja Lennox-Gasteau, 2016).
W rezultacie będą wymagać pomocy w wielu czynnościach i codziennych czynnościach. Tylko niewielki odsetek osób dotkniętych chorobą żyje niezależnie i funkcjonalnie w wieku dorosłym (Genetics Home Reference, 2016).
Inne mniej wspólne funkcje
Oprócz opisanych powyżej objawów przedmiotowych i podmiotowych instytucja Human Phenotype OntologY (2016) odnosi się do szerokiej listy powikłań medycznych, które mogą pojawić się w związku z zespołem Lennoxa-Gastauta (Genetic adn Rare Diseases Information Centre, 2016):
- Strukturalne anomalie mózgu : nieprawidłowości w okołokomorowej istocie białej, amplituda cisterna magna, zanik czołowo-skroniowy, hipoplastyczne ciało modzelowate, makrocefalia.
- Wady rozwojowe czaszkowo-twarzowe : wady zgryzu, obniżony most nosowy, powiększenie dziąseł, wysoki przód, niska implantacja pawilonów słuchowych, obrócone uszy, ptoza.
- Profil neurologiczny: zmienna encefalopatia padaczkowa, postępująca i ciężka niepełnosprawność intelektualna.
- Inne powikłania : dysfagia, refluks żołądkowo-przełykowy, nawracające infekcje dróg oddechowych itp.
Jaki jest typowy przebieg kliniczny zespołu Lennoxa-Gastauta?
Zespół Lennoxa-Gastauta jest uważany za dziecinne zaburzenie padaczkowe, które musi przetrwać przez całe dorosłe życie (Valdivia Álvarez i Marreno Martínez, 2016).
Pierwsze objawy tej patologii pojawiają się częściej między 3 a 5 rokiem życia (David, García i Meneses, 2014).
Niektóre przypadki wystąpienia przed 6 miesiącem można opisać, ale są one związane ze stanem innego rodzaju historii padaczki, takiej jak zespół Westa (Valdivia Álvarez i Marreno Martínez, 2016).
Inne przypadki późnego początku pojawiają się również w pośrednich stadiach dzieciństwa, w fazie dorastania lub dorosłości (Asociación Andaluza de Epilepsia, 2016).
W ponad 80% przypadków zespół Lennoxa-Gastauta objawia się pojawieniem się drgawek (andaluzyjskie stowarzyszenie padaczki, 2016).
Kryzysy te zwykle przybierają postać napadów mioklonicznych, tonicznych lub toniczno-klonicznych. Częstotliwość występowania waha się od 9 do 70 odcinków dziennie (Rey, Encabo, Pizarro, San Martín i López-Timoneda, 2015).
Najczęstsze są kryzysy toniczne, stanowiące do 55% z nich (Rey, Encabo, Pizarro, San Martín i López-Timoneda, 2015).
We wczesnych stadiach tej choroby można również zidentyfikować objawy behawioralne lub neurologiczne. Najczęściej obserwuje się ogólne opóźnienie w rozwoju funkcji poznawczych i psychomotorycznych (Asociación Andaluza de Epilepsia, 2016).
Wraz z rozwojem zespołu Lennoxa-Gastatura kryzysy zwykle ewoluują w kilku kierunkach (Andaluzyjskie Stowarzyszenie Padaczki, 2016):
- Całkowite ustąpienie napadów u około 20% chorych.
- Znaczące zmniejszenie wpływu klinicznego lub nasilenia napadów w 25% przypadków.
- Zwiększenie ciężkości i częstości napadów padaczkowych w ponad 50% zdiagnozowanych przypadków.
W tym drugim przypadku zmiany neurologiczne mają tendencję do utrzymywania się lub nasilania, powodując umiarkowaną lub ciężką niepełnosprawność intelektualną w 80% przypadków (andaluzyjskie stowarzyszenie padaczki, 2016).
Przyczyny
Przyczyny zespołu Lennoxa-Gastauta mogą być bardzo szerokie. Można opisać wiele procesów patologicznych, które zmieniają strukturę i sprawne funkcjonowanie układu nerwowego.
U ponad 70% osób ze zdiagnozowanym zespołem Lennoxa-Gastauta choroba ta ma zwykle rozpoznawalne pochodzenie.
Najbardziej związane z tą patologią odnoszą się do (National Organization for Rare Disorders, 2016):
- Nieprawidłowe lub niedostateczne tworzenie kory mózgowej (dysplazja korowa).
- Wrodzone infekcje
- Urazy czaszkowo-mózgowe.
- Przerwanie lub zmniejszenie podaży tlenu w mózgu (niedotlenienie okołoporodowe).
- Zakażenia układu nerwowego: zapalenie mózgu, zapalenie opon mózgowych, stwardnienie guzowate itp.
Analiza historii choroby pokazuje, że prawie 30% osób dotkniętych chorobą ma wcześniejszą historię zespołu Westa (National Organization for Rare Disorders, 2016):
W przypadkach, w których znaczący przebieg kliniczny nie jest wykrywany, zazwyczaj nie ma historii nieprawidłowości ani patologii mózgu.
W przypadkach z wyraźnym przebiegiem klinicznym, tj. Objawowym, są zwykle związane z medycznymi następstwami zapalenia opon mózgowo-rdzeniowych, epizodami zamartwicy, stwardnieniem guzowatym, urazami głowy, dysplazją korową, guzami mózgu i innymi rodzajami patologii metabolicznych (Campos Castelló, 2007).
Niektórzy badacze i instytucje analizują możliwy wpływ czynników genetycznych na pochodzenie zespołu Lennoxa-Gastauta (Genetics Home Reference, 2016).
Większość przypadków zespołu Lennoxa-Gastauta występuje sporadycznie. Występuje u osób, które nie mają historii rodzinnej chorób padaczkowych (Genetics Home Reference, 2016).
3-30% osób dotkniętych chorobą ma historię rodzinną zgodną z tą patologią. Jednak trwające badania nie zdołały jeszcze powiązać jej przebiegu klinicznego ze specyficznymi mutacjami genetycznymi (Genetics Home Reference, 2016).
Diagnoza
Jak wspomnieliśmy w początkowym opisie, zespół Lennoxa-Gastauta można zidentyfikować klinicznie na podstawie napadów.
Dlatego w obliczu podejrzenia patologii epileptycznej niezbędne jest badanie elektroencefalograficzne aktywności mózgowej (Campos Castelló, 2007).
Ponadto ważne jest przeprowadzenie szerszego badania w celu precyzyjnego określenia jego cech i rozładowania innych rodzajów chorób (Valdivia Álvarez i Marreno Martínez, 2012):
- Skomputeryzowana tomografia osiowa (CAT)
- Magnetyczny rezonans jądrowy (NMR).
- Analiza metaboliczna moczu.
- Badanie hematologiczne.
Na poziomie ogólnym, cechy, które musi spełniać obraz kliniczny osoby dotkniętej chorobą, aby postawić diagnozę zespołu Lennoxa-Gastauta (Valdivia Álvarez i Marreno Martínez, 2012):
- Obecność różnego rodzaju napadów padaczkowych o charakterze ogólnym.
- Brak lub częściowa odpowiedź na leki przeciwpadaczkowe.
- Niepełnosprawność intelektualna, której towarzyszą zmiany i zaburzenia zachowania.
- Aktywność elektroencefalograficzna charakteryzująca się powolnym wzorem fali szczytowej podczas fazy czuwania.
Leczenie
Zespół Lennoxa-Gastauta jest zwykle chorobą przewlekłą, więc osoby dotknięte chorobą będą wymagały leczenia przez całe życie (David, García, Meneses, 2014).
Terapia farmakologiczna
Chociaż większość patologii padaczkowych ma tendencję do pozytywnego reagowania na leki, zespół ten jest zwykle oporny na podawanie leków przeciwpadaczkowych (David, García, Meneses, 2014).
Trwające badania nie wykazały jeszcze lekarstwa na zespół Lennoxa-Gastauta (Fundacja Lennox-Gasteau, 2016).
Początkowo niektóre z najczęściej stosowanych leków to kwas walproinowy, lamotrygina, topiramat, rufinamid, klobazam lub felbamat, przydatne w zwalczaniu napadów padaczkowych (David, García, Meneses, 2014):
- Kwas walproinowy (walproinian) : ten rodzaj leku jest uważany za jeden z pierwszych zabiegów z wyboru. Jest bardzo skuteczny w leczeniu i kontroli różnych form napadów. Zazwyczaj podaje się je indywidualnie (monoterapia). Jeśli nie wykazuje znaczących wyników, może być łączony z innym rodzajem leków, takich jak klobazam, topiramat lub lamotrygina na receptę (National Organization for Rare Disorders, 2016).
- Inne leki : inne leki, takie jak rufinamid, klobazam, topiramat, lamotrygina lub felbamat, mogą pomóc w zmniejszeniu i kontroli aktywności padaczkowej. Jednak niektóre z nich są zwykle związane ze znaczącymi skutkami ubocznymi.
Ten rodzaj leków przeciwpadaczkowych jest zwykle łączony, ponieważ indywidualne podawanie zwykle nie wykazuje znaczącego wpływu na kontrolę objawów padaczkowych (National Institute of Neurological Disorders and Stroke, 2015).
Duża liczba osób dotkniętych chorobą musi poprawić swój stan kliniczny dzięki tego typu podejściu, jednak zazwyczaj ogranicza się to do początkowych momentów (National Institute of Neurological Disorders and Stroke, 2015).
Najczęstszym problemem w zespole Lennoxa-Gastauta jest to, że rozwija się tolerancja na leczenie farmakologiczne i pojawiają się niekontrolowane napady (National Institute of Neurological Disorders and Stroke, 2015).
Terapie dietetyczne
Biorąc pod uwagę ogniotrwałość tego zespołu, można zastosować alternatywne interwencje, takie jak terapie dietetyczne i procedury chirurgiczne (andaluzyjskie stowarzyszenie padaczki, 2016):
W dziedzinie regulacji żywności najczęściej stosowanym podejściem jest przepisanie diety ketogenicznej ( CD ).
Ta interwencja opiera się na regulacji źródeł wkładów energetycznych. Celem jest zastąpienie spożycia węglowodanów lipidami.
Ta rutynowa dieta pozwala na produkcję ciał ketonowych wynikających z metabolizmu spożywanych kwasów tłuszczowych. W rezultacie można uzyskać znaczące zmniejszenie progu padaczki.
Dieta ketogeniczna jest już stosowana w medycynie, jednak ważne jest, aby specjaliści przeprowadzali okresowe kontrole w celu zbadania ich skutków.
Andaluzyjskie Stowarzyszenie Padaczki (2006) stwierdza, że w prowadzeniu badania klinicznego z tego typu dietą 38% uczestników zmniejszyło swój kryzys o ponad połowę.
Ponadto w 7% przypadków przebieg kliniczny uczestników był wolny od napadów.
Procedury chirurgiczne
Interwencja chirurgiczna jest ograniczona do przypadków określonych przez (Asociación Andaluza de Epilepsia, 2016):
- Ciężki przebieg kliniczny.
- Odporność na leczenie farmakologiczne.
Najczęściej stosowanymi procedurami są stymulacja nerwu błędnego i kalosotomia (Asociación Andaluza de Epilepsia, 2016).
Stymulacja nerwów błędnych
Nerw błędny stanowi jedną z gałęzi nerwów lub nerwów czaszkowych. Jego pochodzenie znajduje się w rdzeniu przedłużonym i przebiega przez gardło do różnych narządów wewnętrznych, takich jak wątroba, trzustka, żołądek lub serce.
Wszczepienie podskórnej sondy do stymulacji elektrycznej nerwu błędnego w okolicy podobojczykowej jest stosowane jako technika paliatywna w tego typu zaburzeniach (Neurodidacta, 2012).
Jest to jedna z najbardziej nowatorskich procedur w leczeniu padaczki. Ponad połowie użytkowników udaje się kontrolować napady, zmniejszając je do 50% (Neurodidacta, 2012).
Kalosotomia
Ciało modzelowate jest strukturą zbudowaną z wiązki włókien nerwowych, która łączy dwie półkule mózgowe.
W przypadkach określonych przez (Fundacja Lennoxa-Gastauta, 2016) zalecana jest interwencja chirurgiczna tej struktury poprzez częściową kalosotomię (resekcja przednich trzecich) lub całkowitą (resekcja tylnej części trzeciej).
- Obecność uogólnionych napadów padaczkowych (w obu półkulach mózgowych).
- Powtarzający się kryzys
- Odporność na podawanie leków przeciwdrgawkowych.
Ten rodzaj interwencji może skutecznie zmniejszyć napady w 75% -90% przypadków (Lennox-Gastaut Syndrome Foundation, 2016).
Oprócz tych dwóch technik można również zastosować inne rodzaje podejść, takie jak głęboka stymulacja mózgu lub stymulacja nerwu trójdzielnego (Lennox-Gastaut Syndrome Foundation, 2016):
Głęboka stymulacja mózgu
Wszczepianie elektrod stymulujących w głębokich obszarach mózgu jest metodą stosowaną w leczeniu różnych chorób, takich jak choroba Parkinsona i inne zaburzenia ruchowe.
Wprowadzenie tego typu stymulatorów do przedniego jądra wzgórza stanowi jedną z terapii eksperymentalnych w padaczce.
W Stanach Zjednoczonych klinika Mayo pokazuje, że po 40% stymulacji mózgu nastąpiła znaczna redukcja napadów padaczkowych.
Stymulacja nerwu trójdzielnego
Grupa naukowców z Uniwersytetu Kalifornijskiego w Los Angeles (UCLA) stworzyła równoległy system stymulacji ukierunkowany na podawanie prądów elektrycznych w nerwie trójdzielnym w leczeniu padaczki (NeuroSigma, 2016).
Ta nowa procedura nosi nazwę Monarch Etns System (NeuroSigma, 2016).
Badanie opublikowane przez czasopismo Neurology (2009, 2013) pokazuje, że ponad 40% użytkowników tej eksperymentalnej terapii zdołało zmniejszyć do 50% całkowitą liczbę napadów (NeuroSigma, 2016).
Ponadto terapia ta wykazała korzyści w poprawie nastroju pacjentów, znacznie zmniejszając objawy depresyjne niektórych chorych (NeuroSigma, 2016).
Jakie są prognozy medyczne?
Prognozy medyczne u osób dotkniętych zespołem Lennox-Gasteau są bardzo zmienne (National Institute of Neurological Disorders and Stroke, 2015).
Cierpienie na uporczywe napady padaczkowe i postępujące pogorszenie funkcji poznawczych znacznie ograniczą jakość życia osób dotkniętych chorobą (Lennox-Gasteau Foundation, 2016).
Zazwyczaj nie reagują one korzystnie na klasyczne leczenie farmakologiczne, a częściowe lub całkowite wyzdrowienie jest rzadkością (National Institute of Neurological Disorders and Stroke, 2015).
Śmiertelność tego zespołu sięga 5%. Przyczyny nie są zazwyczaj bezpośrednio związane z samą chorobą, zwykle wynikają z pedagogiki stanu padaczkowego (Campos-Castelló, 2007).
Stan padaczkowy jest stanem medycznym związanym z cierpieniem z powodu długotrwałych napadów (Uninet, 2016).
Ten typ ataków zwykle osiąga czasowość ponad 30 minut i wiąże się z ważnymi powikłaniami: przerwaniem funkcji życiowych, następstwami neurologicznymi, zaburzeniami psychicznymi itp. (Uninet, 2016).
W ponad 20% przypadków śmierć następuje nieuchronnie (Uninet, 2016).
Z drugiej strony utrata przytomności lub nagłe zmniejszenie napięcia mięśniowego towarzyszące niektórym rodzajom napadów jest kolejnym czynnikiem ryzyka, który przyczynia się do zwiększenia śmiertelności w tym zespole (Genetics Home Reference, 2016).
Istotne jest, aby istniał kompleksowy monitoring medyczny i kontrola, zarówno kliniczny przebieg choroby, jak i wtórne komplikacje medyczne.